
NAGS Deficiency Research

©NUCDF (Article reprinted from NUCDF Newsletter Vol XIV, No 1 Spring 2005)
Potential Cure for N-acetylglutamate Synthase Deficiency
with N-carbamylglutamate (Carbaglu®)
By Mendel Tuchman, M.D.
Children’s National Medical Center
Washington, D.C.
New drug offers hope of a normal life to NAGs patients, holds the possibility partial CPS patients could show a response to the drug.
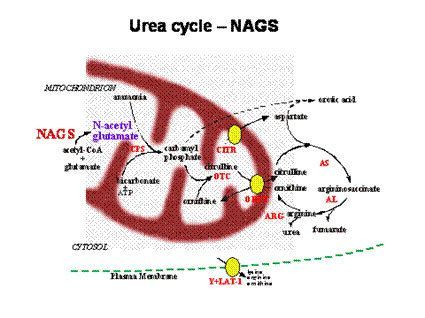
All but one of the genes that encode the proteins required for the urea cycle to function were known for a long time, most since the 1980s. The only gene that remained elusive was the one making the enzyme N-acetylglutamate synthase (NAGS). The NAGS enzyme produces an essential chemical called N-acetylglutamate (NAG) which, if missing, stops the cycle as if CPS or OTC were missing. NAG is essential for CPS (the first enzyme of the cycle) to work. Without it, CPS cannot do its job of detoxifying ammonia. Therefore, a baby born with complete NAGS deficiency will have a clinical picture that is identical to babies born with complete CPS deficiency, including hyperammonemic coma within the first week of life, elevated blood glutamine and low or absent citrulline. The orotic acid in the urine will be low or normal as it is in CPS deficiency.
After researchers in our laboratory identified, cloned and sequenced the human NAGS gene, we understood why it remained elusive all these years; unlike all the other urea cycle genes, the NAGS gene of mammals (including humans) is very different from the NAGS genes of bacteria, fungi and other lower organisms. So, having the sequences of NAGS from these organisms was not as helpful for finding it in humans. Another problem is that unlike all other urea cycle proteins, NAGS is present in the liver in only very small amounts, which made finding the protein and extracting it also very difficult.
Soon after we identified the NAGS gene, we were able to accurately diagnose patients with NAGS deficiency. As in CPS and OTC deficiency, some patients had the neonatal type while others had the late onset type, depending how severe the effect of the mutation is on the function of the protein. Neonatal type mutations generally cause complete deficiency (no activity of the enzyme), while those that allow the enzyme to work at reduce capacity can present at any age, including adulthood. One can justifiably ask, if the role of NAGS is to produce NAG to help CPS work, why can’t we give patients enough NAG (that can be produced in the laboratory quite easily) and cure the patient? After all, each of us probably needs little NAG in the liver to keep the urea cycle going. The answer: if one takes NAG orally or injects NAG, it never gets to the liver, as it is broken down quickly by an enzyme before it can act to help the urea cycle. There is good news, however.
A similar chemical to NAG called N-carbamylglutamate, also activates the CPS enzyme and at the same time is resistant to any breakdown in the body. This chemical, when taken orally, gets to the liver cells and even enters the mitochondria (organelle where CPS is located) and can make CPS work. This chemical is currently not approved for use in the US, but is approved in Europe to treat NAGS patients, and is made by Orphan Europe under the trade name Carbaglu®. A few patients have been treated in Europe and have been reported to respond well to treatment, however, no study has been conducted to investigate this and some of these patients may not have actually had NAGS deficiency, as a completely reliable test for it was not previously available.
Now that we were able to diagnose patients with NAGS deficiency by a reliable DNA test, we could study the effect of Carbaglu® on the urea cycle of patients with this disorder. Orphan Europe graciously agreed to provide us enough drug to study several patients with NAGS deficiency, before and after 3 days of Carbaglu® treatment. Two adult patients with partial NAGS deficiency agreed to be studied before, and following 3 days of Carbaglu® treatment. Using a test developed by Dr. Marc Yudkoff at Children Hospital of Philadelphia, the patients were given a stable isotope to follow how much of the tracer is incorporated into urea, as a measure of how the urea cycle works. We also measured ammonia, urea and amino acids in the blood before and following treatment with Carbaglu®. Before taking the drug, both patients had very little urea production, implying a marked deficiency in detoxifying ammonia. Following 3 days of treatment, the urea production capacity completely normalized, ammonia came down, and the urea in the blood increased markedly. The result of these studies in both patients indicated that N-carbamylglutamate could provide a curing effect of the hyperammonemia from NAGS deficiency. Although such a dramatic effect on a urea cycle disorder is expected only in patients with NAGS deficiency, there is also a possibility that some of the patients with partial CPS deficiency could show a response to the drug, as it could make the residual CPS work better, or it could make the altered CPS protein last longer in these patients. However, this will have to be studied separately. It is also expected that Carbaglu® would effectively treat the hyperammonemia seen in patients with propionic and methylmalonic acidemia (two other inborn errors of metabolism) and investigators in Europe have shown a reduction in the ammonia of these patients following treatment with Carbaglu®. It is less likely that this drug will work for other urea cycle disorders because the enzymes involved are not dependent on the presence of NAG.
Although NAGS deficiency is very rare, we feel that many of these patients remain undiagnosed or some could have been misdiagnosed with CPS deficiency (especially if CPS deficiency was not confirmed on liver biopsy or by DNA analysis). Misdiagnosis as OTC deficiency would be rare, as OTC-deficient patients have elevated urine orotic acid levels. It is important to correctly diagnose NAGS deficiency in patients who may have a clinical picture resembling CPS or OTC deficiency, because specific and effective treatment is now available which would alleviate the need for other drugs, severe protein restriction or liver transplantation. We need to know more about N-carbamylglutamate, such as the optimal dose, efficiency of intestinal absorption and how to maximize it, and minimize any long-term side effects. We hope that future studies will provide answers to these important questions.
Editor’s Note: Dr. Tuchman will present his findings and a patient case study at our 2005 Conference.